信号通路(细胞信号通路详解)
信号通路(细胞信号通路详解)
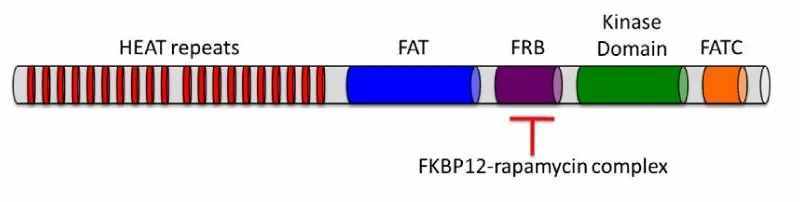
mTOR(哺乳动物雷帕霉素靶标)是一种分子量为289 kDa的丝氨酸/苏氨酸蛋白激酶,属于磷脂酰肌醇3-激酶相关激酶(PIKK)家族。该蛋白由一个催化激酶结构域、一个FRB(FKBP12-雷帕霉素结合)结构域、C-末端附近的一个预测的自抑制结构域(抑制子结构域)、氨基末端多达20个重复的HEAT基序以及FAT(FRAP-ATM-TRRAP)和FATC(FAT C-末端)结构域组成。TOR的C末端与磷脂酰肌醇3-激酶(PI3K)的催化结构域高度同源。TOR蛋白在进化上从酵母到人类都是保守的,人、小鼠和大鼠的mTOR蛋白在氨基酸水平上有95%的同源性。人mTOR基因编码2549个氨基酸的蛋白质,与酵母TOR1和TOR2的序列同源性分别为42%和45%。mTOR在参与控制细胞生长和增殖的信号通路中起中心作用(参考文献1)。
mTOR通路受多种细胞信号的调控,包括有丝分裂生长因子、胰岛素等激素、营养素(氨基酸、葡萄糖)、细胞能量水平和应激条件。PI3K/Akt(v-Akt小鼠胸腺瘤病毒癌基因同源1)信号转导通路是通过mTOR传递信号的主要通路,在介导细胞存活和增殖中起重要作用。通过PI3K/Akt通路的信号是由与细胞膜上的受体结合的生长因子的有丝分裂刺激启动的。这些受体包括IGFR(胰岛素样生长因子受体)、PDGFR(血小板衍生生长因子受体)、EGFR(表皮生长因子受体)和HER家族。来自激活的受体的信号直接传递到PI3K/Akt通路,或者,也可以通过由致癌蛋白RAS激活的生长因子受体激活。RAS是另一个信号转导的中枢开关,而且已证实是MAPK(丝裂原活化蛋白激酶)信号转导通路的关键激活子。胰岛素也可通过IRS1/2(胰岛素受体底物-1/2)激活PI3K/Akt通路。胰岛素结合激活IR(胰岛素受体)酪氨酸激酶,使IRS1或IRS2磷酸化。PI3K通过P85调节亚基中的SH2(Src-Homology-2)结构域与磷酸化IR结合。这种相互作用激活了p110催化亚基。然后,PI3K催化膜结合的PIP2(磷脂酰肌醇(4,5)二磷酸)转化为PIP3(磷脂酰肌醇(3,4,5)-三磷酸)。PIP3然后与Akt的pleckstrin同源结构域结合,通过二聚化和暴露其催化位点而导致Akt的激活。
AKT也可以被PDK-1(磷脂依赖激酶-1)磷酸化和激活。AKT直接磷酸化mTOR。AKT也可能通过TSC1/TSC2(结节性硬化症复合体)的作用间接作用于mTOR。蛋白质TSC1(Hamartin)和TSC2(Tuberin)的物理结合产生了抑制mTOR的功能复合体。最近的证据表明,TSC1/TSC2的抑制作用是通过TSC2失活Ras家族的小GTPase Rheb(RasHomolog Enriched In Brain)实现的。TSC2对Rheb具有GAP(GTPase-Activating Protein)活性,推测TSC1/TSC2复合物通过刺激Rheb的GTP水解来抑制mTOR信号转导。RHEB-GTP激活mTOR。PMA(佛波酯)也可以通过PKC(蛋白激酶-C)和RSK1(核糖体-S6激酶-1)抑制TSC1/2复合体,以及通过PKC激活S6K1而不依赖于Akt而导致mTOR磷酸化。AMPK(AMP(腺苷5‘-单磷酸)激活的蛋白激酶)也可以调节mTOR。AMPK对细胞内AMP(5‘-单磷酸腺苷)/ATP(三磷酸腺苷)比值的升高非常敏感,因此是关键的能量敏感激酶。这一比例的增加促进了上游激酶LKB1的磷酸化和激活,上游激酶LKB1是一种在Peutz-Jeghers综合征中突变的人类肿瘤抑制因子。激活的AMPK反过来磷酸化TSC2(位于与Akt磷酸化的残基不同的残基上),明显促进其激活。这反过来又抑制了mTOR活性的作用(参考文献2,3和5)。
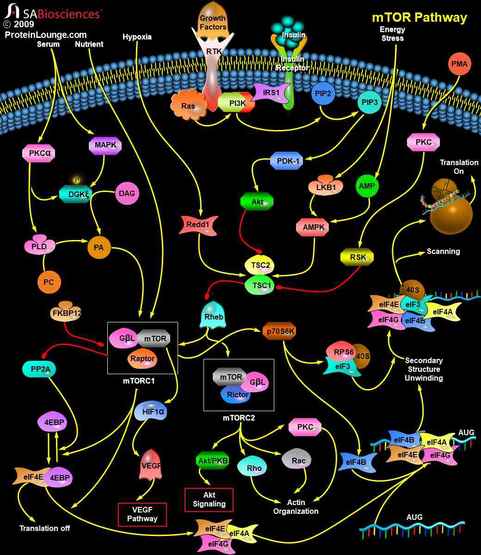
磷脂酸(PA)也能激活mTOR。有三种不同的酶可以产生PA:PLD(磷脂酶-D)、LPAAT(溶血磷脂酸酰基转移酶)和DGK(二酰甘油激酶)。PLD被认为是PA对mTOR信号的主要贡献者。尽管如此,其他产生PA的酶也可以促进mTOR的激活;据报道,LPAAT在一些肿瘤中升高,其过度表达会导致细胞转化。血清刺激导致PLD激活,这与mTOR信号增强相关。血清是有丝分裂原的混合物,通过G蛋白偶联受体(GPCRs)或受体酪氨酸激酶(RTKs)发挥作用。PLD活性随着两种受体类型的刺激而增加。脂类如DAG(二酰甘油)和PA产生于膜结构域中,在那里不同的脂质代谢途径之间保持着密切的联系,从而产生适当的时空反应。PLD和DGK可以并行运行,但它们也可以在单个途径中作为DAG和PAG生成酶。在哺乳动物细胞中,内膜(如高尔基体)产生的PA主要是通过磷脂酰胆碱(PC)的PLD作用产生的。这种PA既可以作为信使,促进囊泡分裂,也可以作为磷酸酶的底物,将PA转化为DAG。因为PC是哺乳动物膜中最丰富的脂质,这个通路是DAG的强大供应者,然后可以用作DGK底物(参考文献4&5)。因此,已经提出了几种机制来解释mTOR是如何受到生长因子和细胞能量水平的调节的。然而,关于mTOR是如何受压力条件调节的,我们知之甚少。两种应激诱导蛋白RTP801/Redd1和RTP801L/Redd2通过mTOR有效地抑制信号转导。RTP801和RTP801L作用于AKT下游和TSC2上游,以抑制mTOR功能。另一种mTOR抑制剂是雷帕霉素。当与其细胞受体FKBP12(FK506结合蛋白-12)络合时,雷帕霉素直接与TOR结合以抑制下游信号(参考文献1、6和7)。
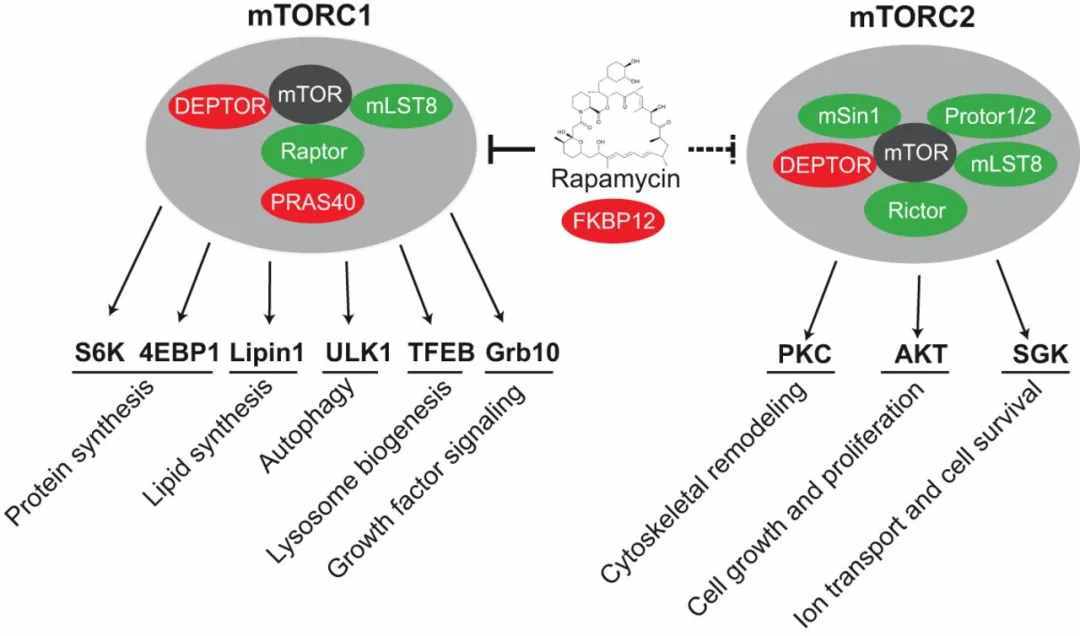
mTOR的激活会导致几个下游靶点的磷酸化。蛋白质mTOR要激活其信号级联,必须形成三元复合体mTORC1(mTOR复合体-1)和mTORC2(mTOR复合体-2)。雷帕霉素敏感的mTORC1控制着几条共同决定细胞质量(大小)的通路。雷帕霉素不敏感的mTORC2控制肌动蛋白细胞骨架,从而决定细胞的形状。mTORC1(和可能的mTORC2)是多聚体,尽管会绘制为单体。mTORC1是由mTOR、RAPTOR(mTOR调节相关蛋白)和G-BetaL(G-蛋白β亚基样蛋白)组成的三元复合物。另一方面,mTORC2复合物由mTOR、G-BetaL和Rictor组成。mTOR下游研究最清楚的效应器是两条信号通路,它们平行作用,控制mRNA的翻译。激活的mTOR介导eIF4EBP1(真核翻译起始因子-4E结合蛋白-1)和核糖体蛋白p70S6K或S6K1(S6激酶)的磷酸化。4EBP1(又称PHAS1)是一种能抑制eIF4F(真核细胞起始因子-4)复合物活性的小分子蛋白。在非磷酸化状态下,4EBP1/PHAS1与eIF4F复合物的mRNA帽结合亚基eIF4E(真核翻译起始因子-4E)紧密结合,从而抑制eIF4E启动蛋白质合成的活性。mTOR使4EBP1磷酸化,降低其与eIF4E的亲和力,使两种蛋白解离。然后eIF4E能够与eIF4F的其他成分结合,这些成分包括大支架蛋白eIF4G(真核翻译起始因子-4-γ)、依赖三磷酸腺苷的RNA解旋酶eIF4A(真核翻译起始因子-4A)和eIF4B(真核翻译起始因子-4B),形成活性复合物。这种复合体促进了帽子依赖蛋白的翻译。净效应是具有5’-非翻译区的mRNAs子集的翻译增加,这些非翻译区通常编码与细胞周期中的增殖反应和从G1期到S期的转换相关的蛋白质。这些mRNA包括编码c-Myc、CCND1(Cyclin-D1)和鸟氨酸脱羧酶的那些。Cyclin-D1与CDK4结合,形成Rb(视网膜母细胞瘤蛋白)磷酸化所需的复合物,促进细胞周期和DNA复制。剥夺生长因子或抑制mTOR导致4EBP1去磷酸化,并与eIF4E重新结合,随后帽特异性翻译减少。mTOR还可能通过调节PP2A(蛋白磷酸酶-2A)的活性间接影响4EBP1的磷酸化状态。mTOR下游的第二个主要效应因子是S6K1丝氨酸/苏氨酸激酶。在接收到PI3K/Akt通路介导的增殖上游信号后,mTOR磷酸化并激活S6K1。反过来,S6K1磷酸化并激活40S核糖体S6蛋白,促进40S核糖体亚基募集到激活的翻译多聚体中。特别地,具有5’-top(5’-T末端寡嘧啶)序列的mRNAs的翻译被增强。这些具有5’-TOP的mRNAs主要编码核糖体蛋白、延伸因子和IGF-II(胰岛素样生长因子-II)。S6K1的去磷酸化减少了蛋白质翻译系统各组成部分的合成,导致蛋白质合成的显著减少。mTORC1还通过磷酸化HIF1Alpha(缺氧诱导因子-1-Alpha亚单位)来调节VEGF(血管内皮生长因子)(参考文献8,9和10)。
除了对翻译的影响外,mTOR还通过调节RNA聚合酶I和III来调节蛋白质的合成,这两个聚合酶负责核糖体和转运RNA的转录。在适当的生长信号如IGF1的存在下,mTOR与PI3K和MAPK通路一起调控Pol I介导的核糖体RNA的转录。也有证据表明,mTOR可能通过影响调控Rb上游CDK的Cyclin-D1和p27的稳定性和表达来调节Rb的磷酸化状态,从而对聚合酶产生作用。mTORC2可能通过一个小的Rho型GTPase和PKC向肌动蛋白细胞骨架发出信号。此外,mTORC2以生长因子依赖的方式控制激活的、GTP结合的rac1的形成。mTORC2还控制PKC-α(protein Kinase-C-Alpha,PKC-Alpha)的磷酸化和活化。mTOR作为增殖信号转导的中枢调控因子,是治疗肿瘤的理想靶点。通过对许多信号转导途径的广泛阐明,mTOR激酶参与了整合外部信号和内部信号的关键事件,协调细胞的生长和增殖。mTOR接收指示转录和翻译机制是否应该上调的信号,然后有效地将这些信号传送到适当的途径。在许多癌症类型中,通过mTOR传递信号的信号通路的多个组成部分是失调的。开发mTOR抑制剂是治疗以mTOR信号通路失调为特征的恶性肿瘤的合理治疗策略(参考文献9&11)。
参考文献:
1. Wullschleger S, Loewith R, Hall MN. TOR signaling in growth and metabolism. Cell. 2006 Feb 10;124(3):471-84. PubMed ID:16469695
2. O'Reilly KE, Rojo F, She QB, Solit D,Mills GB, Smith D, Lane H, Hofmann F, Hicklin DJ, Ludwig DL, Baselga J, RosenN. mTOR inhibition induces upstream receptor tyrosine kinase signaling andactivates Akt. Cancer Res. 2006 Feb 1;66(3):1500-8. PubMed ID: 16452206
3. Jozwiak J, Jozwiak S, Grzela T,Lazarczyk M. Positive and negative regulation of TSC2 activity and its effectson downstream effectors of the mTOR pathway. Neuromolecular Med.2005;7(4):287-96. PubMed ID: 16391386
4. Avila-Flores A, Santos T, Rincon E,Merida I. Modulation of the mammalian target of rapamycin pathway bydiacylglycerol kinase-produced phosphatidic acid. J Biol Chem. 2005 Mar18;280(11):10091-9. PubMed ID: 15632115
5. Han S, Khuri FR, Roman J. Fibronectinstimulates non-small cell lung carcinoma cell growth through activation ofAkt/mammalian target of rapamycin/S6 kinase and inactivation ofLKB1/AMP-activated protein kinase signal pathways. Cancer Res. 2006 Jan1;66(1):315-23. PubMed ID: 16397245
6. Cortot A, Armand JP, Soria JC. PI3K-AKT-mTORpathway inhibitors. Bull Cancer. 2006 Jan 1;93(1):19-26. PubMed ID: 16455502
7. Ellisen LW. Growth control under stress:mTOR regulation through the REDD1-TSC pathway. Cell Cycle. 2005Nov;4(11):1500-02. Epub 2005 Nov 1. PubMed ID: 16258273
8. Fumarola C, La Monica S, Alfieri RR,Borra E, Guidotti GG. Cell size reduction induced by inhibition of themTOR/S6K-signaling pathway protects Jurkat cells from apoptosis. Cell DeathDiffer. 2005 Oct;12(10):1344-57. PubMed ID: 15905878
9. Lang CH, Frost RA. Endotoxin disruptsthe leucine-signaling pathway involving phosphorylation of mTOR, 4E-BP1, andS6K1 in skeletal muscle. J Cell Physiol. 2005 Apr;203(1):144-55. PubMed ID:15389631
10. Fiano V, Ghimenti C, Imarisio S,Silengo L, Schiffer D. PAkt, cyclin D1 and p27/Kip.1 in glioblastomas with andwithout EGFR amplification and PTEN mutation. Anticancer Res. 2004Sep-Oct;24(5A):2643-7. PubMed ID: 15517868
11. Choo AY, Blenis J. TORgeting oncogeneaddiction for cancer therapy. Cancer Cell. 2006 Feb;9(2):77-9. PubMed ID:16473275